
自2015年8月18日《国务院关于改革药品医疗器械审评审批制度的意见》发布以来,提高药品审批标准和推进仿制药质量一致性评价的相关政策,拔高了化学仿制药的审批标准,引发国内两千多家化学仿制药生产厂家的“生存保卫战”。
药品审批标准的提高,主要是指仿制药由当时的“仿已有国家标准的药品”标准,调整为“仿与原研药品质量和疗效一致的药品”标准。于是,未申报的仿制药一律要以原研药品作为参比制剂,确保新批准的仿制药质量和疗效与原研药品一致。对已经批准上市的仿制药,则按与原研药品质量和疗效一致的原则,分期分批进行质量一致性评价。
相关配套政策发布时间集中在2016年,2016年仿制药申报一度跌入历史新低。那么,2017年仿制药申报到底是什么情况?数据的背后又透露出哪些新的问题?
2017申报临床、BE备案、一致性评价补充申请数据:
BE启动数量大提升
2017年新申报临床的化学药品中,已确定注册分类的有318个受理号,其中:新3类(仿制境外上市但境内未上市原研药品的药品)仅2个,为新疆特丰药业的水合氯醛直肠用溶液和咪达唑仑口服液;新4类(仿制境内已上市原研药品的药品)有1个,为云南龙海天然植物药业的丙泊酚中/长链脂肪乳注射液;新1类有271个,占比68%;新2类有44个。由此可见,新3类申报大幅度下滑。
已公开的BE备案登记号数趋势,也可侧面反映BE研究“宽进严出”之后,BE研究启动数量仍有可能逐步回复到2015年前申报的全盛期:2016年BE研究备案数为25条,2017年为385条,2018年前2个半月(截至2018年3月21日)为84条。
一致性评价的补充申请开放在2017年下半年,目前已有109个受理号,其中获批的受理号数22个,占比超过20%。瑞舒伐他汀钙片和苯磺酸氨氯地平片是补充申请受理号数最多的两个产品。瑞舒伐他汀钙片不是国家要求第一批完成一致性评价289目录的产品。虽然国家要求分期分批进行一致性评价,但大多数企业最终会选择市场潜力大的产品提前进行一致性评价以争取政策红利。
新注册分类前的受理药品,选哪条路径?
“新4类”通道更快?
对于药品注册分类改革前受理的药品注册申请,则有两条通路:
一是继续按照原规定进行审评审批,在质量一致性评价工作中逐步解决与原研药品质量和疗效一致性问题,按旧化学药注册分类申报的2017年以后获批的仿制药生产批文中,都有在一定期限内完成一致性评价的要求。
另一条路就是企业自愿申请按与原研药品质量和疗效一致的新标准审批,当时政策承诺会设立绿色通道,按新的药品注册申请收费标准收费,加快审评审批。截至2018年3月20日,如表1所示,共有3个产品以新4类获批。那么这三个产品有无加快申报呢?
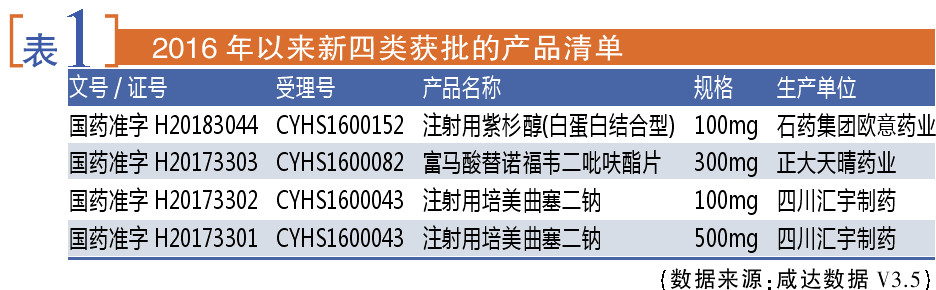
四川汇宇制药的注射用培美曲塞二钠2016年以新4类申报,并且以“已在欧盟递交注册申请,获得英国MHRA、GMP认证”为由获得优先审评,最终在同年获批。苏州特瑞药业的同名竞品2015年以旧6类申报,排队直到2017年8月才获得资料发补的通知。
富马酸替诺福韦二吡呋酯片方面,成都倍特药业、安徽贝克生物制药和齐鲁制药以旧注册分类3.4类报产。2012年安徽贝克生物制药和江苏正大天晴药业分别完成BE研究,首先以旧6类报产;2013年齐鲁制药报产;浙江南洋药业和珠海联邦制药中山分公司2014年报产。历经2015年临床自查核查后,首先获批的是旧3.4类报产的成都倍特;齐鲁排第二,安徽贝克排第三,都是旧6类报产的受理号;最后则是正大天晴以新4类申报的受理号,该受理号还以“抗艾滋病药物”为由获得优先审评。目前,成都倍特和齐鲁已经获得一致性评价标识;正大天晴以新4类获批,理论上也属于通过一致性评价的产品,无需再补充申请就获得一致性评价标识。但CDE“通过一致性评价信息”栏目中目前并没有收录新4类的产品,而没有获得一致性评价标识将有可能会影响正大天晴在采购招标中的竞争位置。
注射用紫杉醇(白蛋白结合型)仅石药集团欧意药业和江苏恒瑞医药股份两家,以“临床急需、市场短缺”为由获得优先审评,两个受理号都是仿制药上市(完成BE研究)阶段。石药欧意受理号较前以新4类注册申报,江苏恒瑞以旧6类注册申报,最终石药欧意赢得了首仿。
综上所述,目前已获批的新4类产品通常也获得优先审评,从注射用培美曲塞二钠案例来看,新4类绿色通道的排队还是比旧6类要快。
参比制剂的可获得性,是否已经完全解决?
还有细节问题待解
仿制药要求质量和疗效与原研药品一致,原研对照药品(即参比制剂)就变得非常关键。但是参比制剂的购买如何合规,成为各企业研制过程中需要面对的现实问题。
2016年7月发布的《总局关于研制过程中所需研究用对照药品一次性进口有关事宜的公告》(2016年第120号)对“以中国境内药品注册为目的的研究中用于对照药品的制剂或原料药”和“以仿制药质量和疗效一致性评价为目的的研究中用于对照药品的化学药品制剂或原料药”,都要求“对符合条件的药物研制过程中所需对照药品,可予以一次性进口”。
2017年8月《总局关于仿制药质量和疗效一致性评价工作有关事项的公告》进一步放开了对“以仿制药质量和疗效一致性评价为目的的研究中用于对照药品的化学药品制剂或原料药”的参比制剂确定和获得途径。企业除了可以通过申报一次性进口申请及进口备案、通关等程序来获得参比制剂,还可通过其他方式获得参比制剂,在提交一致性评价资料时,仅需在资料中提供购买凭证、产品包装及说明书等材料,或以其他适当方法证明参比制剂真实性即可。
“以中国境内药品注册为目的的研究中用于对照药品的制剂或原料药”和“以仿制药质量和疗效一致性评价为目的的研究中用于对照药品的化学药品制剂或原料药”,是两类不同的情况。前者主要针对未上市在申报的仿制药,后者主要针对国内已上市的仿制药。而后者参比制剂的优化措施能否惠及前者,暂无相对应的法规公告。
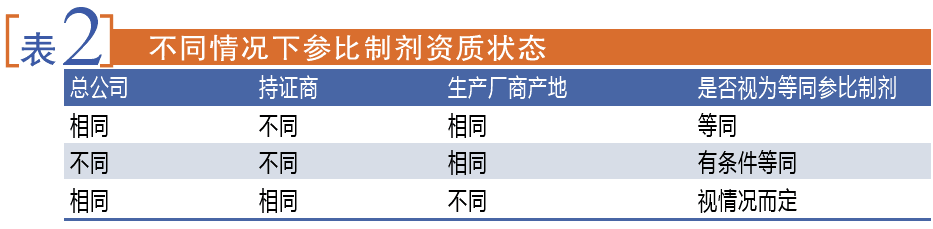
如表2所示,2017年8月《已发布参比制剂有关事宜说明》反映了CFDA最关注的是原研药品生产厂商产地。如果原研药在国内并没有再继续销售,原研进口又被CFDA列入参比制剂目录。国内仿制药企业在美国购买参比制剂,可能面临当时原研在国内申报进口的生产厂商产地可能与美国在销的生产厂商产地不一致的问题,除非国内仿制药企业能提供适宜的证据证明不同产地产品的处方、生产工艺和产品质量相同,才能视为等同。但是,国内仿制药企业基本不能提供,于是该产品就有可能因为参比制剂买不到该规格,从而不能在国内上市或通过一致性评价。
小结<<<
已上市产品的一致性评价标准,与未上市产品“仿与原研药品质量和疗效一致”的注册标准理应一致,但笔者在日常工作时发现,两类产品之间的政策仍有一些不同点。
预计一致性评价的补充申请和仿制药的申报在2018年将保持增长,新4类的报产受理号在2018年有可能会出现报复性增长。